Cystic Fibrosis Symptoms and Treatment – Key Signs, Diagnosis & Breakthroughs
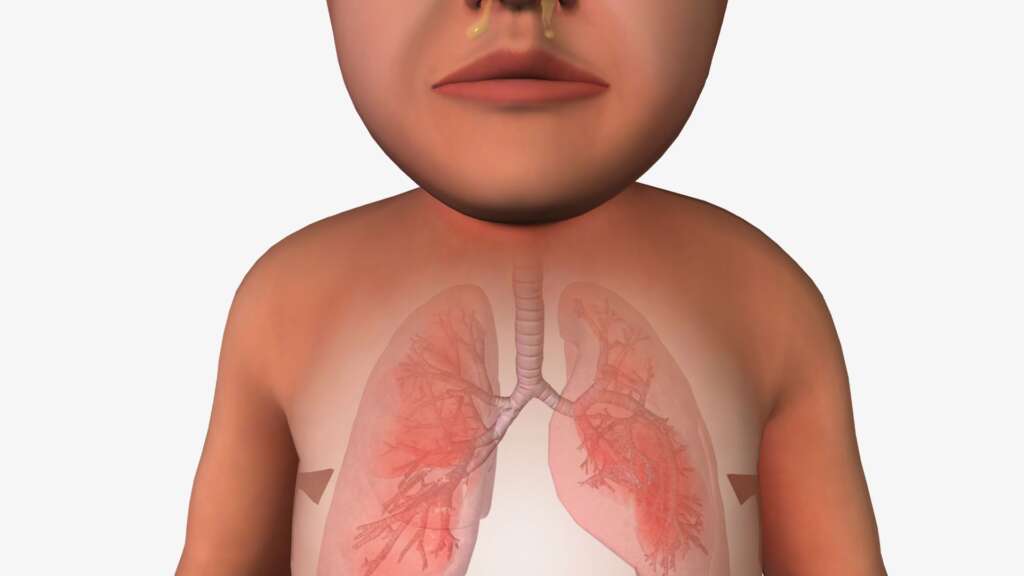
| Highlights Of Cystic Fibrosis
- Cystic fibrosis is a genetic condition that causes thick, sticky mucus buildup in organs like the lungs and digestive system.
- Cystic fibrosis occurs due to the mutation in the Cystic Fibrosis Transmembrane conductance Regulator(CFTR) gene inherited from both parents.
- Symptoms of cystic fibrosis vary and can include chronic cough, wheezing, greasy stool, lung infection, and slow growth.
- The Sweat Test is used to diagnose Cystic Fibrosis. It checks for the amount of chloride in one’s sweat.
- While there’s no cure for Cystic Fibrosis, treatment focuses on symptom management to ensure clear airways through medications, physical therapy, and even surgery.
Cystic Fibrosis – What Is It?
Cystic Fibrosis (CF) is a genetic disease that causes thick mucus and other fluids to build up and clog different parts of the body, including the lungs and digestive systems. Understanding cystic fibrosis symptoms and treatment matters early, as the disease leads to several health issues such as chronic cough, trouble breathing, lung infection, greasy stool, and poor growth. According to the American Lung Foundation, about 70000 people are suffering from CF in the world, and about 30000 people are in the United States. [1]
Causes and Risk Factors for Cystic Fibrosis
CF is caused by an abnormal Cystic Fibrosis Transmembrane conductance Regulator gene — a CFTR gene mutation. Cystic fibrosis is inherited from both parents: if you inherit the abnormal gene from both of your parents, you will develop CF; if you inherit it from only one parent, you will not have CF yourself — you will simply be a carrier of the gene. All ethnic groups can be affected by CF; however, it is more common in people of Northern European descent.
Symptoms of Cystic Fibrosis
CF can cause different symptoms at different times and with varying severity. Most people start experiencing symptoms as babies or young children, while some may not develop until they reach teens or adults. CF mainly affects the respiratory and digestive systems.
-> Respiratory Symptoms
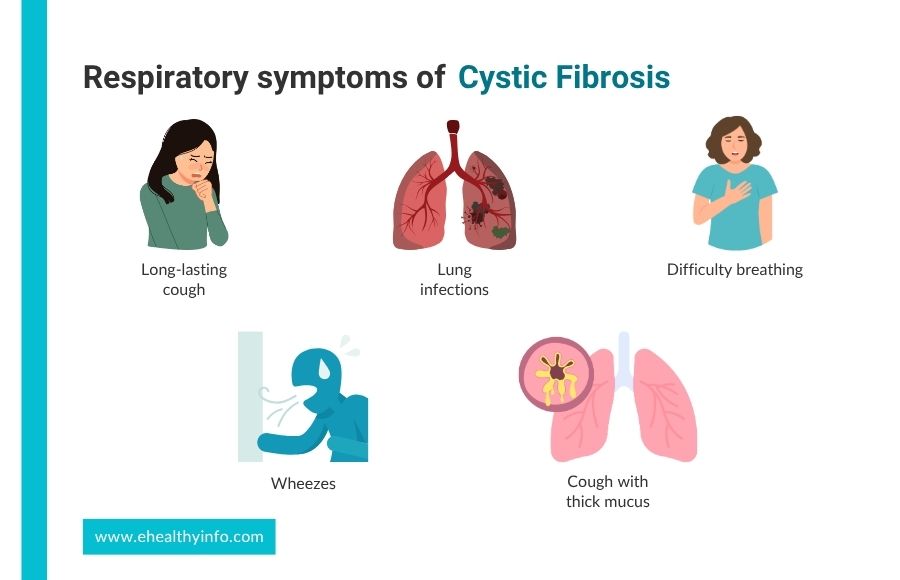
- Long-lasting cough, often with thick mucus, which is difficult to clear.
- Frequent lung infections, such as bronchitis and pneumonia
- Difficulty in breathing
- breathing that sounds like whistling (wheezing)
-> Digestive Symptoms
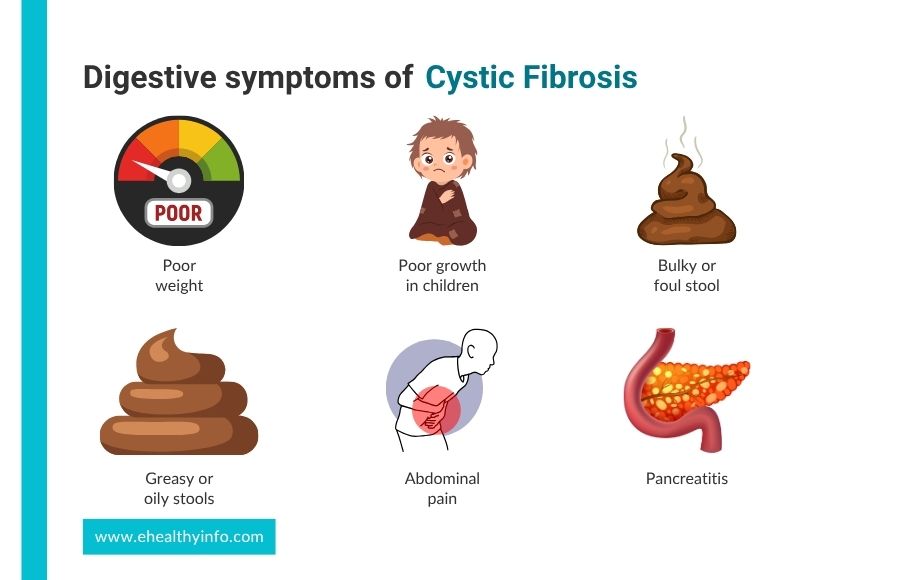
- Poor weight gain or poor growth in children.
- Frequent, bulky, foul-smelling stools.
- Abdominal pain and discomfort.
- Greasy or oily stools.
- Pancreatitis (inflammation of the pancreas).
-> Other Symptoms
- Salty-tasting skin (due to abnormally high salt levels in sweat).
- Dehydration and electrolyte imbalances.
Diagnosis of Cystic Fibrosis
The diagnosis of CF typically involves: [3]
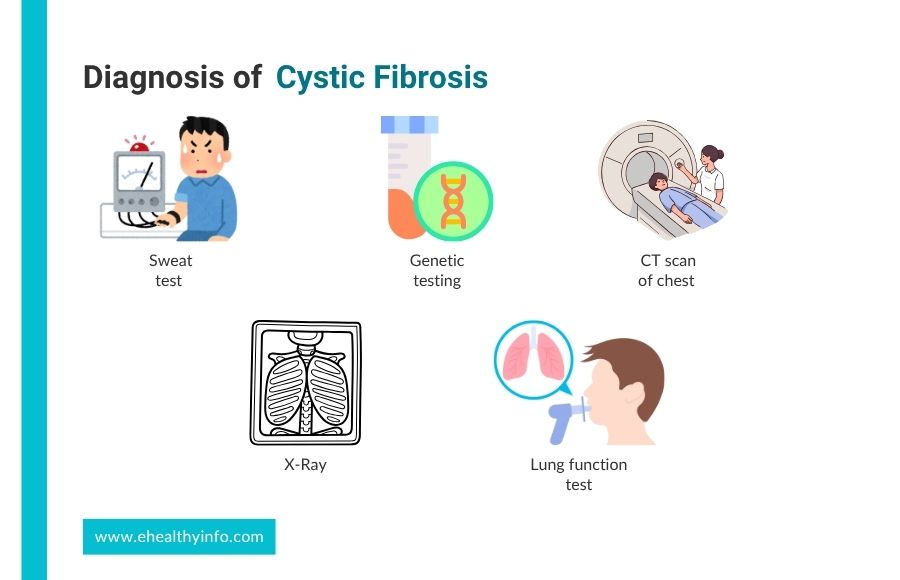
1. Sweat Test for Cystic Fibrosis
The sweat test for cystic fibrosis is one of the most common diagnostic tests, where your doctor checks the amount of chloride in your sweat.. People with CF can have about two to five times the normal amount of chloride in their sweat. In this test, the skin is stimulated to produce sweat, which is then absorbed in a special collector and analyzed.
2. Genetic Testing
Genetic testing determines whether certain changes in the CFTR gene are present. It helps confirm if you have CF and identify the specific genetic changes causing it. This test might be done if a newborn screening shows a possible problem or if the doctor suspects CF based on symptoms.
3. X-rays
Chest X-rays or other imaging studies may be ordered to evaluate lung function and assess for signs of lung damage or infection, which are common in individuals with CF.
4. Computed Tomographic (CT) scan of the chest
CT scans of the chest provide information regarding structural changes in the lungs and aid in planning treatment. CT scans provide detailed and precise images of the chest and abdomen and help assess lung health, detect complications, and evaluate sinus disease. In individuals with CF, CT scans can help identify abnormalities such as bronchiectasis, mucus plugs, lung infections, pneumothorax, and pleural effusion.
5. Lung Function Tests
The most popular lung function test uses a device known as a spirometer. You inhale fully and then force the breath into the spirometer’s tip. This will help your doctor see how well you can breathe and monitor your lung health.
6. Other Laboratory Tests
Your doctor may perform additional tests to evaluate pancreatic function, nutritional status, and other CF-related organ systems. These tests may include pulmonary function tests, pancreatic enzyme tests, liver function tests, and blood tests to assess vitamin and mineral levels.
Complications of Cystic Fibrosis
Some of the complications of cystic fibrosis are given below:
1. Respiratory Complications
CF can lead to chronic lung infections, inflammation, and progressive lung damage. Bronchiectasis is a common complication of CF, resulting in recurrent lung infections, coughing, wheezing, and shortness of breath. Chronic respiratory complications can lead to respiratory failure and the need for oxygen therapy or lung transplantation.
2. Gastrointestinal Complications
CF can cause pancreatic insufficiency, characterized by malabsorption of nutrients, poor growth, weight loss, and vitamin deficiencies. Cystic fibrosis-related diabetes may also develop due to damage to the pancreas. Meconium ileus, a blockage of the intestine in newborns, is another gastrointestinal complication of CF.
3. Nutritional Complications
Malabsorption of nutrients due to pancreatic insufficiency can result in malnutrition, poor growth, and failure to thrive in individuals with CF
4. Sinopulmonary Complications
Chronic sinusitis, nasal polyps, and sinus infections are common sinus-related complications of CF, resulting from impaired mucus clearance and chronic inflammation of the nasal passages and sinuses.
5. Reproductive Complications
CF can affect reproductive health in both males and females. Congenital absence of the vas deferens is a common complication of CF in males, leading to infertility due to blockage of the sperm ducts. In females, CF-related complications may include delayed puberty, irregular menstrual cycles, and decreased fertility.
6. Liver Complications
CF-related liver disease, including hepatobiliary abnormalities, liver cirrhosis, and liver failure, can occur in individuals with CF. Liver complications may result from bile duct obstruction, inflammation, and scarring, leading to impaired liver function and complications such as jaundice, portal hypertension, and ascites.
7. Bone Complications
Individuals with CF may be at increased risk of developing osteoporosis and osteopenia due to factors such as malnutrition, chronic inflammation, and corticosteroid use
Treatment of Cystic Fibrosis
The treatment of CF is comprehensive and aims to manage the symptoms, prevent complications, and improve the overall quality of life.[2] Here are the key components of CF treatment:
1. CFTR modulators
CFTR modulator drugs, such as ivacaftor (Kalydeco), lumacaftor/ivacaftor (Orkambi), and tezacaftor/ivacaftor (Symdeko), target underlying genetic defects. These modulator drugs can improve the function of the CFTR protein and may benefit individuals with specific CFTR mutations. The eligibility for these medications depends on your genotype and specific drug approvals in your region.
2. Supportive Medications
These medications aim to improve lung function, facilitate airway clearance, address nutritional deficiencies, and prevent or manage complications.
Common supportive medications include:
- bronchodilators like albuterol to help open the airways and improve breathing
- mucolytics like Pulmozyme to thin mucus and promote clearance from the airways, pancreatic enzyme replacement therapy to aid digestion and nutrient absorption
- fat-soluble vitamin supplements to address deficiencies
- antibiotics to treat and prevent lung infections
3. Physiotherapy
Physiotherapy is one of the most important aspects of managing CF and mainly consists of airway clearance techniques. These techniques include clapping on the front and back of your chest, using positive expiratory pressure (PEP) therapy, high-frequency chest wall oscillation devices, and postural drainage of sputum. These techniques are typically performed multiple times daily and may vary based on your age and lung function. Besides airway clearance techniques, regular exercise and physical activity can further aid airway clearance and improve lung function and overall fitness.
4. Lung Transplantation
It is a treatment option reserved for individuals with CF who have advanced lung disease and are experiencing respiratory failure despite medical management. Lung transplantation involves replacing one or both diseased lungs with healthy donor lungs obtained from a deceased organ donor. While lung transplantation can improve quality of life and prolong survival in individuals with CF, it is associated with risks and complications, including rejection, infection, and side effects of immunosuppressive medications. Additionally, lung transplantation may not be suitable for all individuals with CF, and careful consideration of the risks and benefits is essential in determining candidacy for transplantation.
Role of Nutrition in Cystic Fibrosis
Nutritional assessment and support from your dietitian are crucial to ensure proper growth and weight maintenance [4]. The best diet for someone with cystic fibrosis is high in calories and fat, with added vitamins and salt. Here are the key dietary tips for CF patients:
1. High Calorie and High Fat Diet
Due to the difficulty digesting and absorbing nutrients and lipids from your diet, you require 1.5 to 2 times as many calories as those without the condition. A smart strategy will be to increase the number of calories in a snack you already had or add an additional 300-500 calories. Pancreatic Enzyme Replacement Therapy (PERT): Pancreatic enzyme replacement therapy (PERT) is a crucial part of CF management, helping to aid digestion and absorption of nutrients by replacing deficient pancreatic enzymes. PERT enables individuals with CF to better utilize nutrients from food and optimize nutritional status.
2. Vitamin and Mineral Supplements
People with CF usually require fat-soluble vitamin supplements (A, D, E, and K) to address the deficiencies of impaired fat absorption. Supplementation helps prevent deficiencies and ensure adequate intake of essential vitamins necessary for various physiological functions.
3. Salt Intake
There is no specific amount mentioned about salt. However, people with CF who play or exercise outside in hot weather might need to add 1/8 teaspoon of salt to 1 1/2 cups of a sports drink, such as Gatorade. Infants with CF should also get 1/8 teaspoon of salt daily until they are six months old and 1/4 teaspoon daily afterward.
4. Feeding Tube
CF impedes digestion, which makes it difficult to absorb nutrients from meals. Your physician might advise using a feeding tube to provide additional nourishment. This tube may be surgically installed in your belly, or it may be a temporary tube that is guided from your nose to your stomach. The tube doesn’t stop mouthfeeding and can provide extra calories at any time of day or night.
Preconception Counseling and Newborn Screening
Preconception counseling plays a pivotal role in individuals with CF planning to start a family. This counseling offers insights into the risk of passing CF to offspring, reproductive options, and prenatal testing. Genetic counseling assists individuals in comprehending their carrier status, the likelihood of having a child with CF, and available family planning options like prenatal testing, preimplantation genetic diagnosis (PGD), and adoption. [5]
Newborn screening (NBS) for CF aims to identify infants with CF shortly after birth for early diagnosis and intervention. The screening involves testing a blood sample from the newborn’s heel for elevated levels of immunoreactive trypsinogen (IRT), often elevated in infants with CF. Infants who screen positive undergo further diagnostic testing, like sweat chloride and genetic testing, to confirm the diagnosis. Early diagnosis through NBS facilitates prompt initiation of CF-specific treatments and interventions, ultimately enhancing outcomes and quality of life for individuals with CF.
Although there is no cure yet, recognizing cystic fibrosis symptoms and treatment options early — from the sweat test to CFTR modulators, airway clearance, and nutrition support — can meaningfully improve quality of life and survival.
Questions To Ask Your Doctor
- Are all my kids at risk of getting cystic fibrosis?
- Are there any exercises that can improve my lung function?
- What are the conditions that I need to be alert to living with cystic fibrosis?
- When do I need a lung transplant, and how would I know if I am eligible for it?
- Can Cystic Fibrosis recur after my Lung Transplantation?
References
-
American Lung Association. (n.d.). Learn about cystic fibrosis. https://www.lung.org/lung-health-diseases/lung-disease-lookup/cystic-fibrosis/learn-about-cystic-fibrosis
- Cleveland Clinic. (n.d.). Cystic fibrosis. https://my.clevelandclinic.org/health/diseases/9358-cystic-fibrosis
- Mayo Clinic. (n.d.). Cystic fibrosis: Diagnosis and treatment. https://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/diagnosis-treatment/drc-20353706
- Johns Hopkins Cystic Fibrosis Center. (n.d.). Nutrition. https://hopkinscf.org/clinical-care/nutrition/
- UpToDate. (n.d.). Cystic fibrosis carrier screening. https://www.uptodate.com/contents/cystic-fibrosis-carrier-screening


