Sickle Cell Disease – Symptoms and Treatments Demystified
What is Sickle Cell?- Understanding The Disease
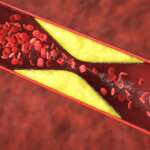
Sickle cell disease (SCD) comprises a collection of hereditary blood cell disorders impacting hemoglobin, the protein responsible for transporting oxygen throughout the body. This condition affects over 100,000 individuals in the United States and around 20 million worldwide. In healthy individuals, red blood cells possess a disc-like shape and flexibility, enabling smooth movement through blood vessels. However, SCD causes the red blood cells to adopt a crescent or “sickle” shape. These abnormal cells lose their ability to bend and move easily, leading to potential blockages in blood flow to various parts of the body.
Sickle Cell Disease and Its Risk Factors
SCD predominantly affects individuals of African ancestry or those identifying as Black in the United States. Around 1 in 13 Black or African American babies are born with sickle cell trait, while approximately 1 in every 365 African American babies are born with SCD. Moreover, the condition also affects individuals from Hispanic, Southern European, Middle Eastern, or Asian Indian backgrounds.
Sickle Cell Disease Symptoms
SCD is a genetic condition that you’re born with, but its effects might not appear until around 5 or 6 months. SCD symptoms can vary from person to person and change over time. Some of the most common signs that are seen in SCD are:
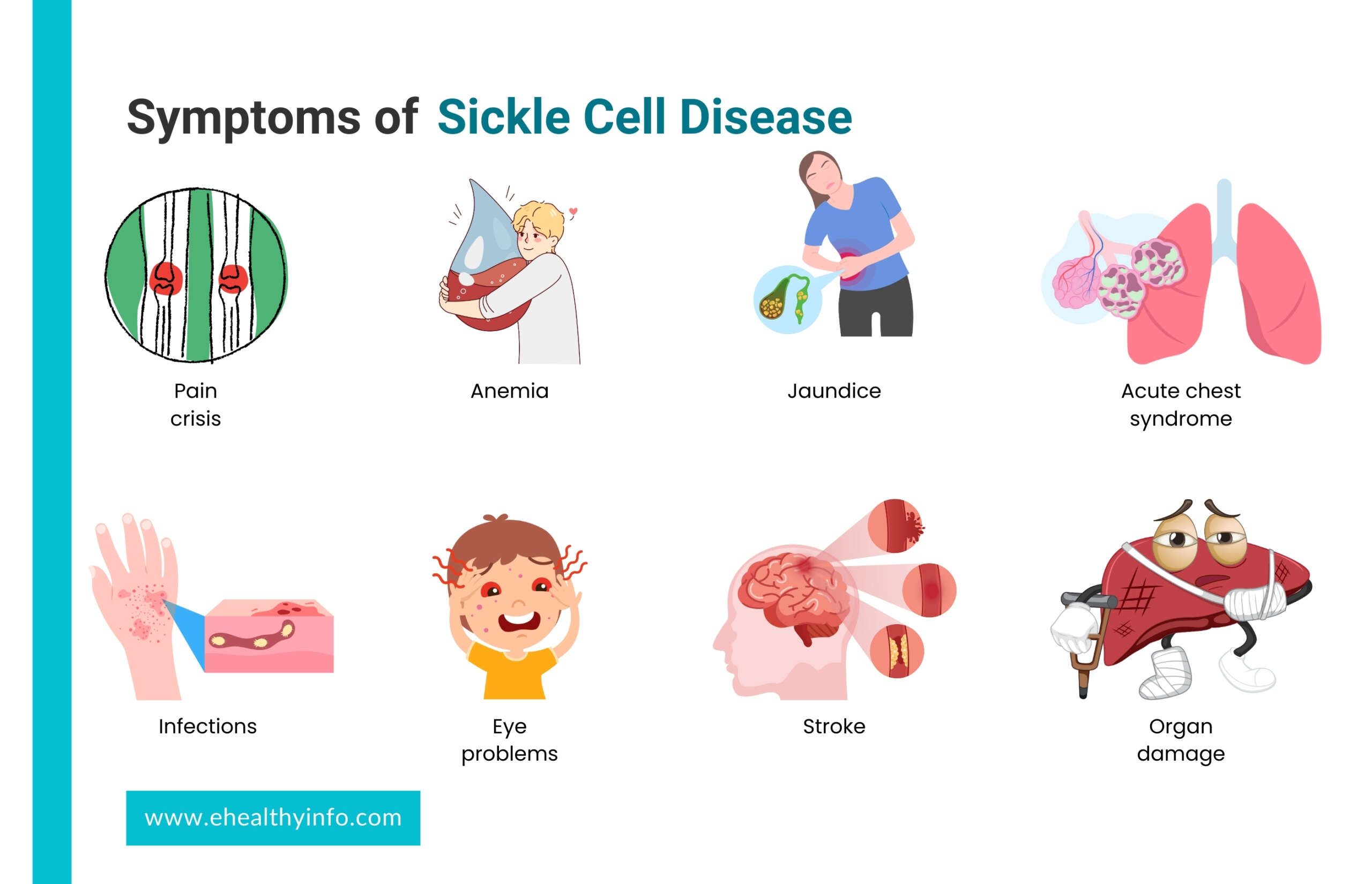
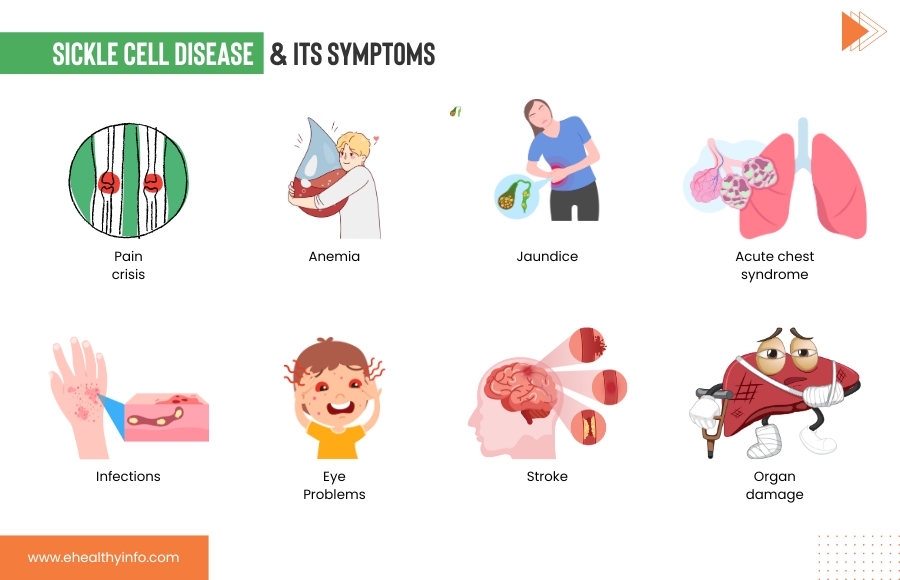
- Pain Crisis (Sickle Cell Crisis): This is one of the hallmark symptoms of SCD. Sickle cells can block blood flow through small blood vessels, causing episodes of severe pain. These episodes are also called the pain crises. These crises can occur anywhere in the body but often affect the bones, joints, chest, and abdomen.
- Anemia: The abnormal shape of sickle cells makes them fragile and prone to breaking apart, leading to a shortage of red blood cells. This can result in anemia, characterized by fatigue, weakness, and paleness.
- Jaundice: Sickle cells have a shorter lifespan than normal red blood cells, leading to a buildup of bilirubin in the blood. This can cause a yellowish tint to the skin and eyes, a condition known as jaundice.
- Infections: Sickle cells can impair the immune system’s ability to fight off infections, making individuals with SCD more susceptible to bacterial infections, particularly those caused by certain types of bacteria, such as Streptococcus pneumoniae.
- Organ Damage: The repeated episodes of blocked blood flow can damage various organs, including the spleen, kidneys, liver, and lungs. This can lead to organ dysfunction and long-term complications.
- Stroke: Sickle cells can also block blood vessels leading to the brain, causing a stroke. Symptoms may include sudden weakness, numbness, confusion, and difficulty speaking or walking.
- Acute Chest Syndrome: This is a serious condition that resembles pneumonia. It occurs when sickle cells block the blood vessels in the lungs, causing chest pain, cough, fever, and difficulty breathing.
- Priapism: It is a condition where individuals experience a prolonged and painful erection that can last for more than 4 hours. It happens due to blocked blood vessels in the penis and requires immediate management.
- Eye Problems: SCD can affect the blood vessels in the eyes, potentially leading to vision problems and even blindness.
Diagnosis of sickle cell disease
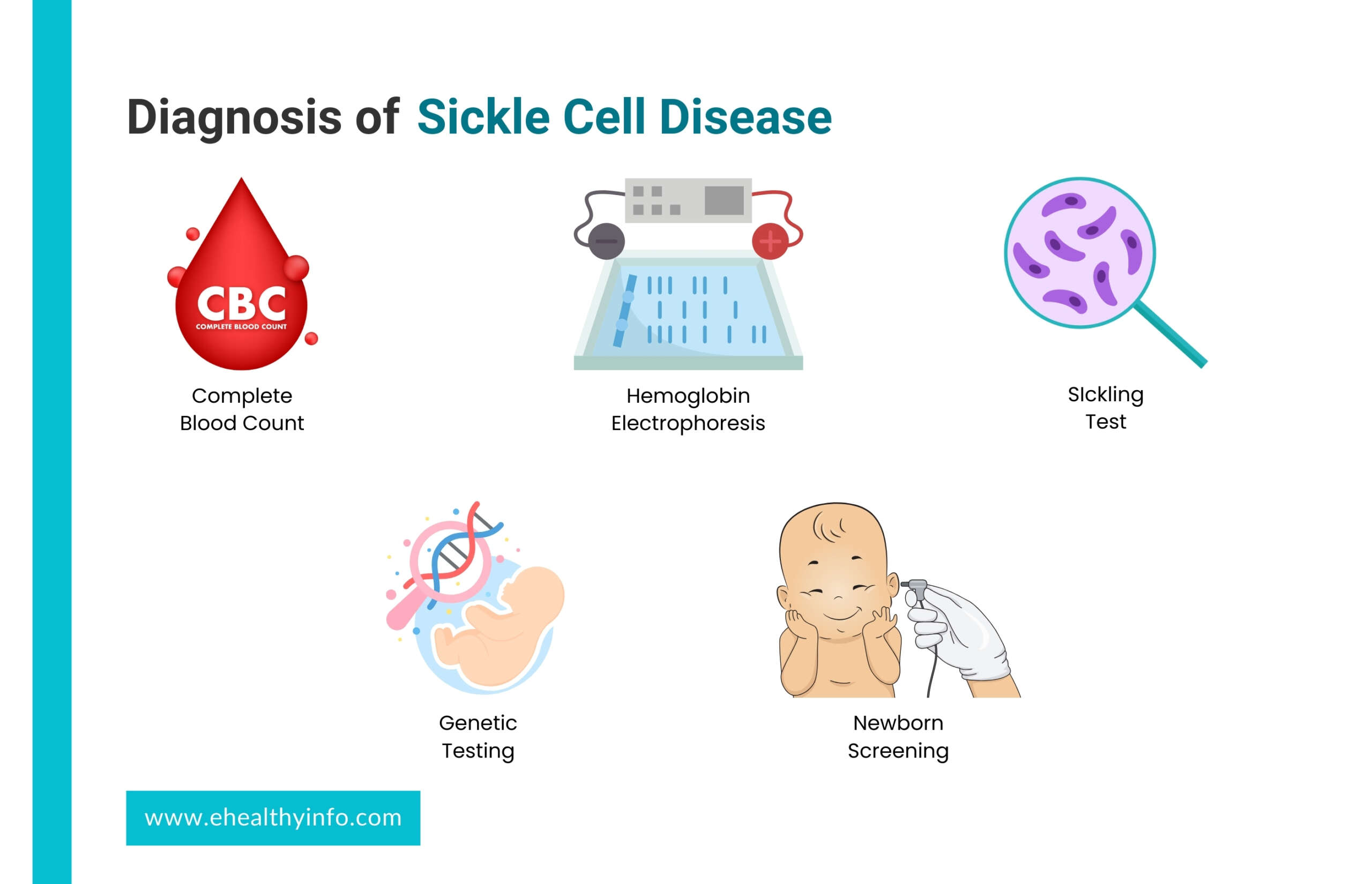
1. Blood Tests: Blood tests play a crucial role in diagnosing SCD. These tests include:
- Complete Blood Count (CBC): This measures the number of red blood cells, white blood cells, and platelets in your blood. People with SCD often have fewer red blood cells and higher levels of reticulocytes (young red blood cells).Hemoglobin
- Electrophoresis: This test identifies the types of hemoglobin present in your blood. In SCD, there is a higher hemoglobin S (HbS) level, responsible for the characteristic sickling of red blood cells, which can be seen with the help of electrophoresis.
2. Sickling Test: In this test, a sample of your blood is exposed to low oxygen levels to observe how your red blood cells behave. In SCD, the cells will take on a characteristic sickle shape under these conditions.
3. Genetic Testing: Genetic tests can confirm the presence of abnormal hemoglobin genes that cause SCD. This is particularly important for carrier screening and prenatal testing. Hemoglobin gene mutations are identified through DNA analysis.
4. Newborn Screening: In many countries, including the United States, newborns are screened for SCD in routine newborn screening programs. This helps identify babies with SCD early on so that appropriate care can be provided. During a newborn screening, drops of blood from a hill prick are collected on a special type of paper. The screening helps to determine whether your baby has a sickle cell trait or is a carrier.
Treatments of Sickle Cell Disease
The treatments of SCD aim to manage symptoms, prevent complications, and improve the quality of life for individuals with the condition. Treatment approaches can vary based on the severity of the disease and the specific symptoms experienced. Here’s an overview of treatment strategies for SCD:
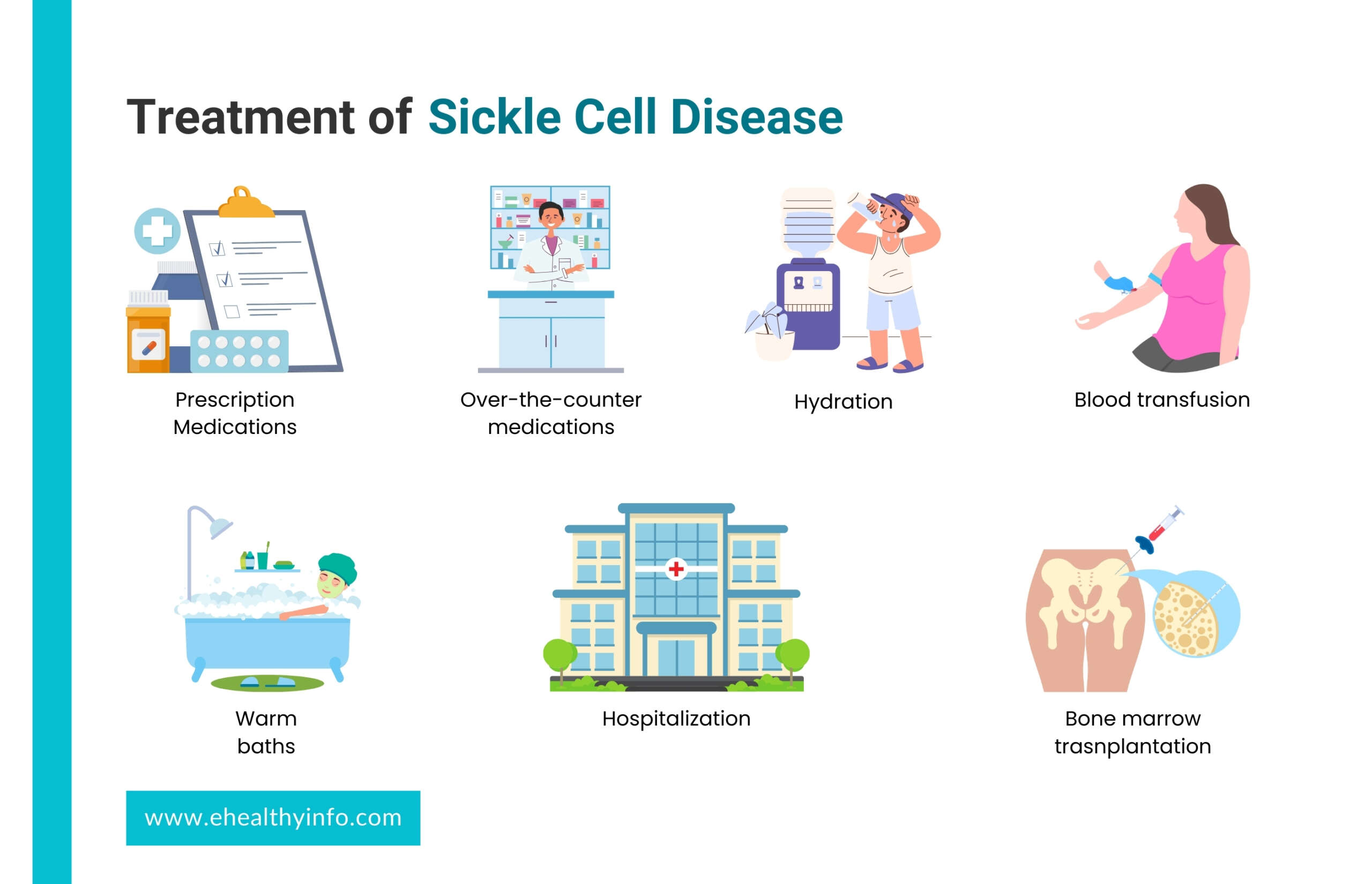
1. Pain Management
- Pain is a common symptom of SCD, often resulting from blocked blood vessels and tissue damage. Pain crises can be managed with pain medications, both over-the-counter and prescription medications.
- Hydration and warm baths can also help alleviate pain during a crisis.
- For severe pain, hospitalization and stronger pain medications may be necessary.
2. Blood Transfusions
- Regular blood transfusions may be recommended for individuals with severe anemia, acute chest syndrome, or other complications.
- Transfusions help increase the number of healthy red blood cells in circulation.
3. Hydration
You need to stay well-hydrated to prevent the formation of sickle cells and reduce the risk of pain crises.
FDA-Approved Treatment
- Hydroxyurea: Hydroxyurea is a well-established medication for SCD. By increasing the production of fetal hemoglobin (HbF), it helps inhibit the sickling of red blood cells and prolong their lifespan. This can reduce pain crises, episodes of acute chest syndrome, and the need for blood transfusions. While hydroxyurea offers substantial benefits for many individuals with SCD, its usage requires careful monitoring due to potential side effects such as changes in blood counts and liver function.
- L-Glutamine (Endari): L-Glutamine is an oral medication approved for treating SCD. This amino acid enhances antioxidant levels in red blood cells, decreasing oxidative stress and improving their function. L-glutamine can help mitigate the frequency of pain crises and hospitalizations, particularly in children and adults with SCD. Its unique formulation as a powder mixed with a liquid makes it convenient for daily use.
- Voxelotor (Oxbryta): Voxelotor is an oral medication that offers a promising approach to managing SCD. By increasing the oxygen affinity of hemoglobin, it prevents the sickling of red blood cells and reduces hemolysis. This results in improved anemia and a decrease in pain crises. Voxelotor is approved for individuals aged 12 and older with SCD.
4. Crizanlizumab (Adakveo)
Crizanlizumab is a monoclonal antibody infusion designed to prevent vaso-occlusive crises in SCD. By blocking the interaction of sickle cells with blood vessel walls, it reduces the frequency and severity of painful episodes. Administered through intravenous infusions, crizanlizumab has shown promise in improving the quality of life for people with SCD.
5. Bone marrow or stem cell transplantation
Bone marrow also known as hematopoietic stem cell transplantation (HSCT), presents a potential curative avenue for SCD. This procedure involves substituting the patient’s malfunctioning bone marrow with healthy donor marrow or stem cells to produce normal red blood cells. Though promising, HSCT is complex and involves risks, including graft-versus-host disease, infection, and organ damage. It is generally reserved for individuals with severe SCD and high-risk complications. Many people who have the disease do not have a relative who is a close match to be a donor. HSCT necessitates meticulous patient selection, close monitoring, and specialized post-transplant care.
Prevention of Sickle Cell Disease
Preventive measures for SCD aim to minimize complications, improve quality of life, and enhance overall health. These strategies focus on managing symptoms, reducing the frequency of pain crises, and preventing infections. Key preventive measures include:
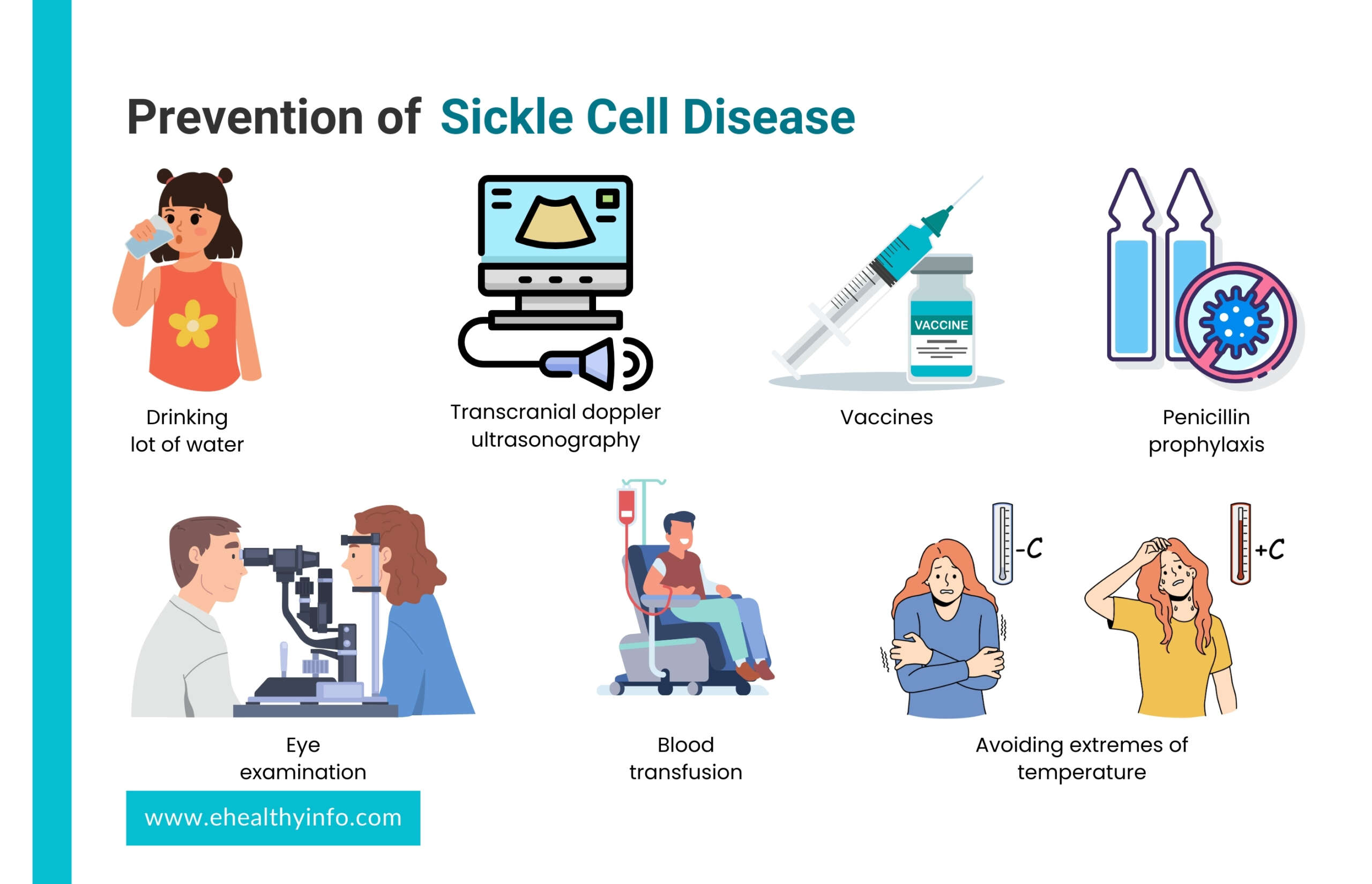
Lifestyle behavior
- Drink a lot of water; it helps prevent sickle cell formation.
- Avoid extremes of temperature, as very cold and hot temperatures can trigger pain crises.
Interventions & Medical Screenings to Reduce SCD Complications
- Vaccines: You or your child with SCD should get all the vaccinations as scheduled, particularly against infections like influenza, pneumonia, and meningococcal disease. Vaccination will help to prevent the disease that can trigger SCD complications.
- Penicillin prophylaxis: Children with SCD need to be given daily doses of penicillin or other antibiotics that help to reduce the risk of bacterial infection in the early days of their growth and development.
- Eye examination: It is recommended that individuals with SCD should receive annual eye examinations to check for possible retinal damage, which is a common complication of SCD.
- Transcranial Doppler ultrasonography (TCD), a specialized type of examination, can be used to detect children at risk for stroke.
- To prevent severe anemia, the patient may be treated with blood transfusions.
When To See Your Doctor?
It is crucial to recognize when immediate medical attention is needed, as SCD can sometimes lead to severe and life-threatening health issues. If you notice the following symptoms or complications, it’s essential to seek medical help or call 9-1-1 without delay:
- Severe Anemia Symptoms: These include extreme tiredness, shortness of breath, dizziness, or irregular heartbeat. Conditions like splenic sequestration or aplastic crises can lead to severe anemia, posing serious risks.
- Fever: If someone with SCD has a fever above 101.3°F (38.5°C), seeing a healthcare provider promptly is critical. Antibiotics may be necessary, and hospitalization might be required in some cases.
- Acute Chest Syndrome Symptoms: Symptoms encompass chest pain, coughing, fever, and difficulty breathing. Admission to a hospital becomes necessary, where treatments like antibiotics, oxygen therapy, or blood transfusions may be administered.
- Stroke Warning Signs: These include sudden weakness, numbness on one side of the body, confusion, and trouble with speaking, seeing, or walking. These signs would indicate a stroke requiring immediate medical intervention.
- Priapism: In cases where an erection lasts for more than 4 hours, it’s advised to visit a hospital. Consultation with a hematologist and a urologist is crucial, as they specialize in blood conditions and male reproductive/urinary system issues, respectively
Questions For Your Doctor
- What type of sickle cell disease do I have?
- What are the common symptoms and complications associated with my type of SCD?
- Is there anything I can do at home to improve my health?
- Are there specific triggers I should be aware of?
- When should I go to the emergency room?
- Can my children get this from me?
- What are my family planning options?
References
- Sickle cell disease | sickle cell anemia (no date) MedlinePlus. Available at: https://medlineplus.gov/sicklecelldisease.html
- (What is sickle cell disease? (2023) Centers for Disease Control and Prevention. Available at: https://www.cdc.gov/ncbddd/sicklecell/facts.html
- https://www.nhlbi.nih.gov/health/sickle-cell-disease/treatment